
Em estudo clínico inédito nos EUA, primeiro autista recebe dose de terapia genética
Um marco importante para a ciência aconteceu nesta semana: pela p...
 Reprodução / Depositphotos
Reprodução / Depositphotos
Um gene, muitas histórias: o que a ciência descobriu sobre autismo e sete mais condições
Nova descoberta genética desafia a forma de classificar 8 transtornos mentais e abre caminho pa...
 Depositphotos
Depositphotos
Fórum debate cenário da terapia gênica no brasil e lança documentário em Brasília
Na próxima sexta-feira, dia 08.ago.2025, será realizado o Fóru...
 Depositphotos
Depositphotos
Novo estudo revela 4 subtipos de autismo e abre caminho para diagnósticos mais precisos
Um estudo publicado nesta semana na revista...
 Reprodução / Universidade Federal de Ouro Preto
Reprodução / Universidade Federal de Ouro Preto
Seminário de genética no autismo é anunciado em MG
A Universidade Federal de Ouro Preto (UFOP) e o Instituto Diferente anunciam o I Simpósio de Genét...


 Depositphotos
Depositphotos
Syngap1 traz mais respostas sobre a relação entre autismo e deficiência intelectual
Recentes descobertas científicas ampliaram significativamente o ent...
 Depositphotos
Depositphotos
Esperma paterno pode estar ligado ao autismo, diz pesquisa
A causa do autismo é...
 Reprodução / Tismoo
Reprodução / Tismoo
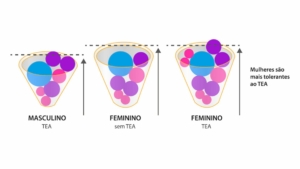
Meninos: ainda são a maioria no TEA
Essa foto é muito intrigante para mim, bem como o modelo que ela representa. Mesmo já tendo escrit...
 Depositphotos
Depositphotos
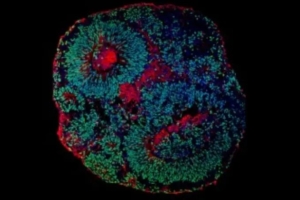
Maior estudo genômico do autismo revela 134 genes ligados ao TEA
Liderado pelo SickKids, o estudo sequenciou os genomas completos...
 Muotri Lab
Muotri Lab
Estudo abre novas possibilidades de tratamento para forma sindrômica de autismo
Liderado por brasileiros, grupo descobre mecanismo causador da síndrome de Pitt-Hopkins, disfun...